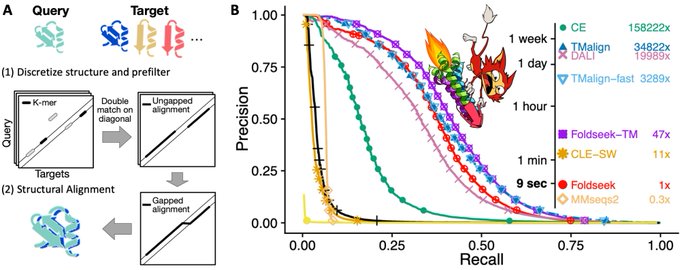
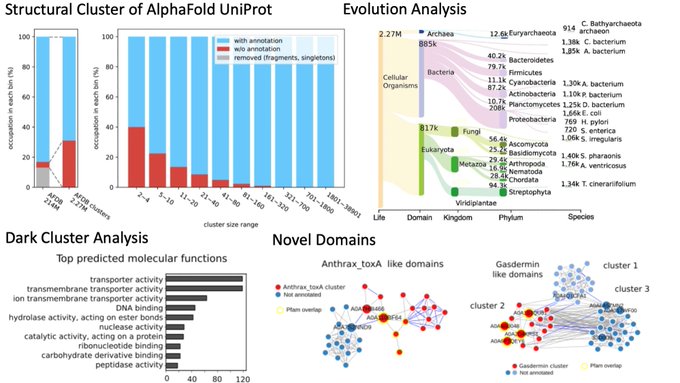
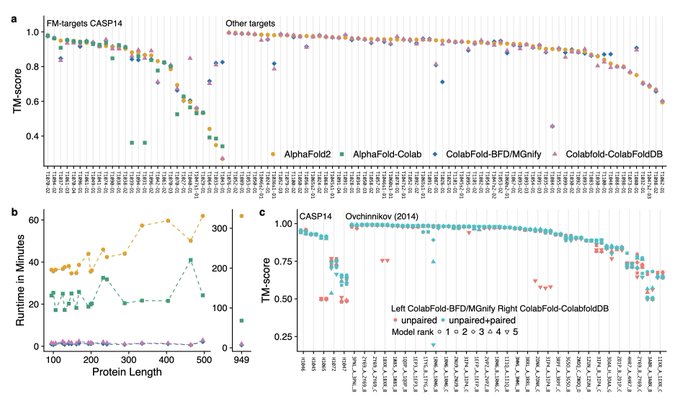
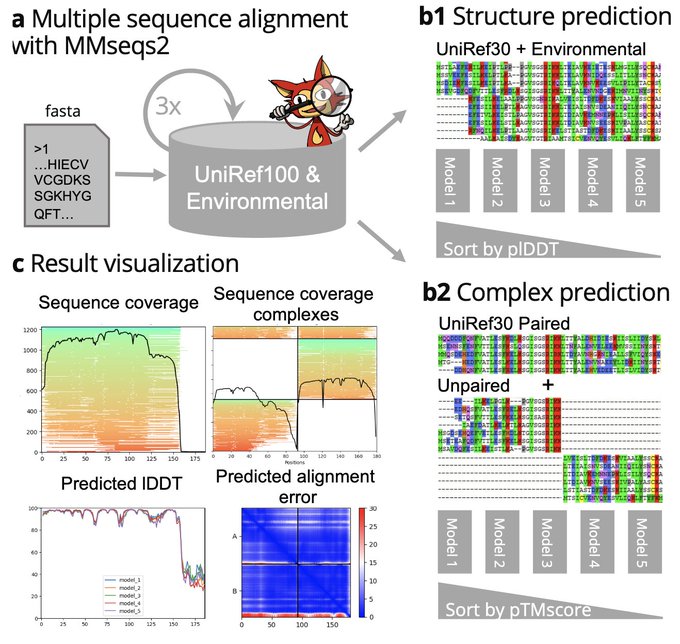
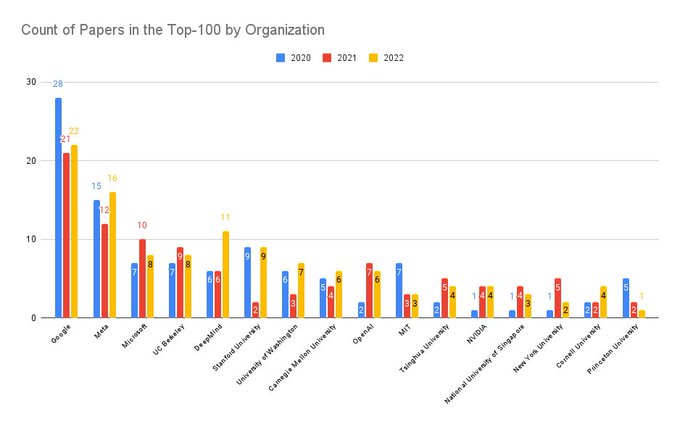
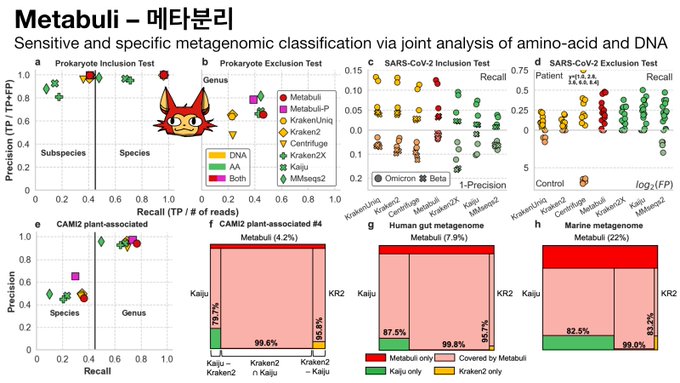
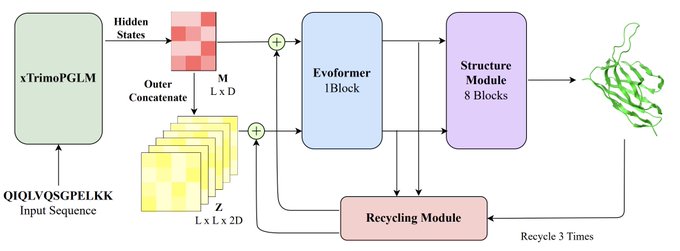
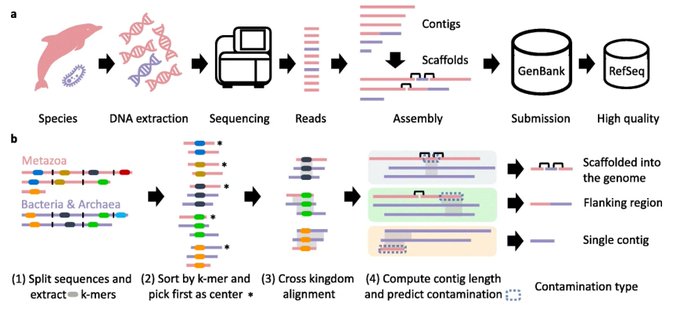
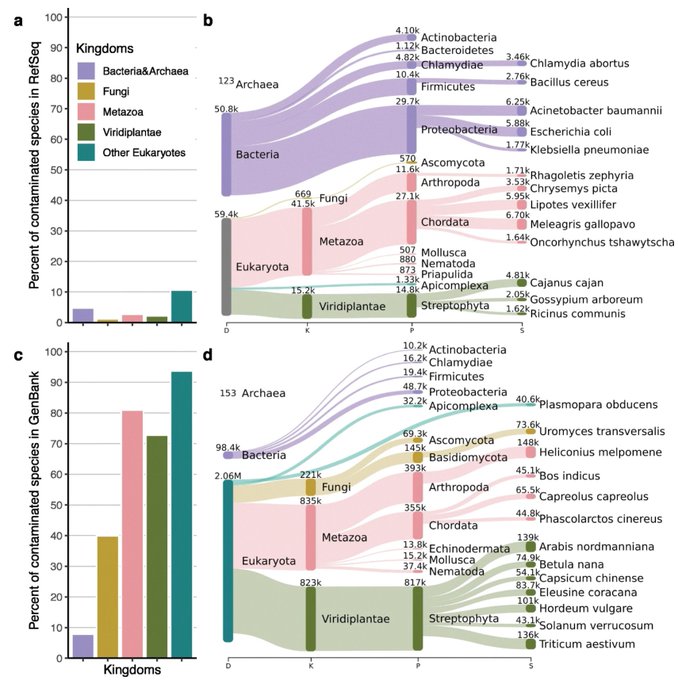
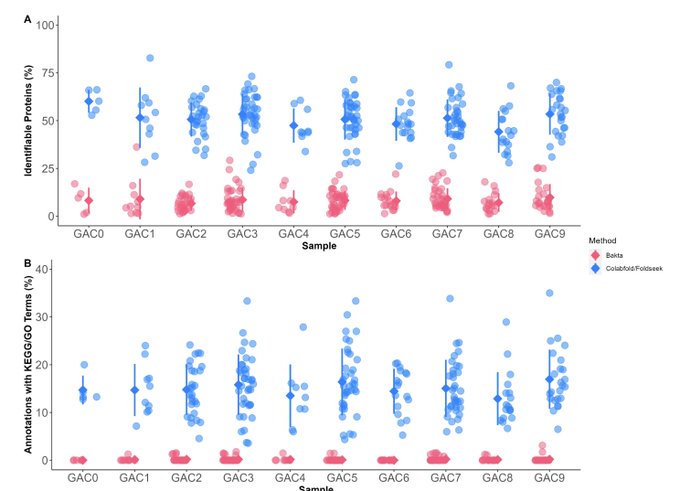
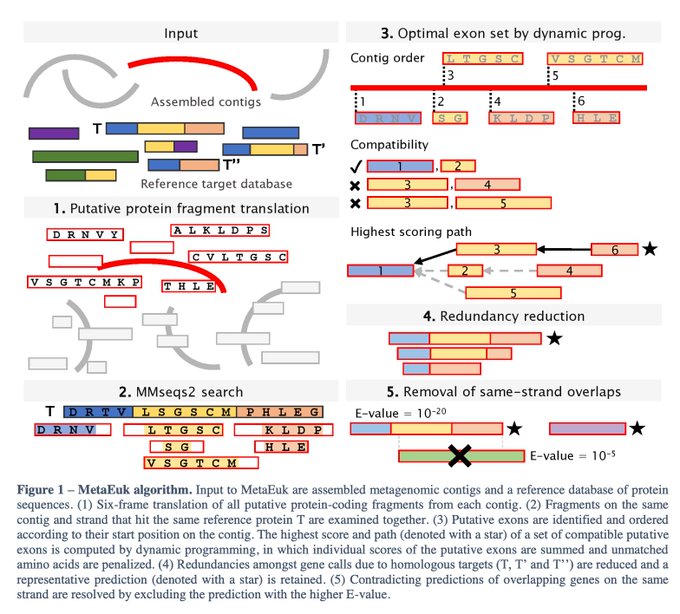
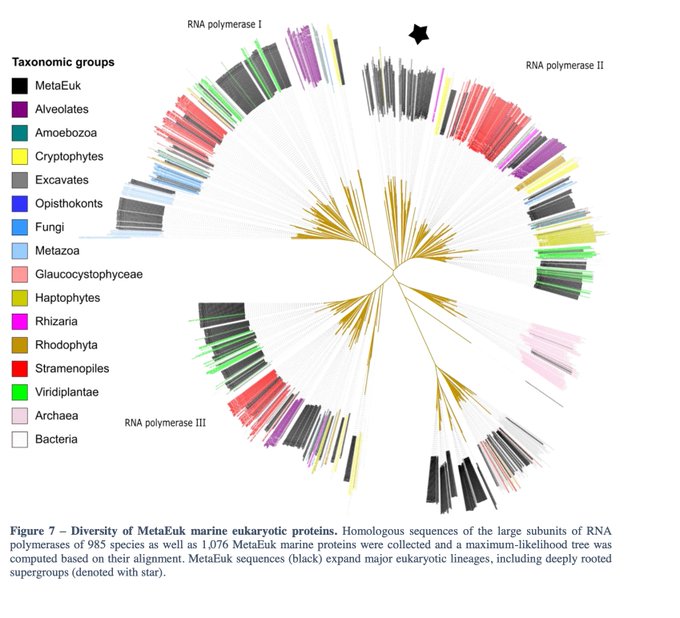
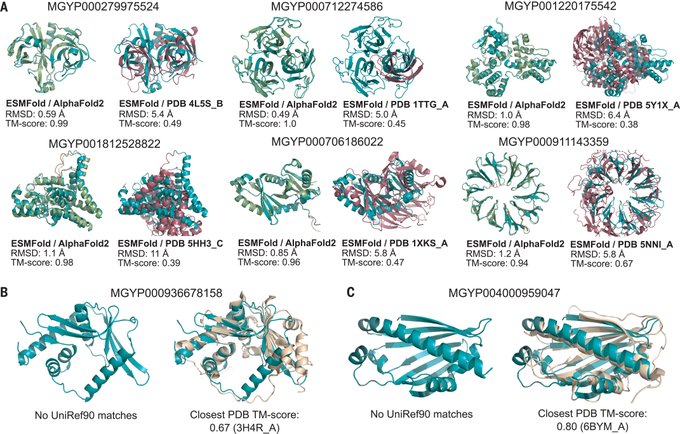
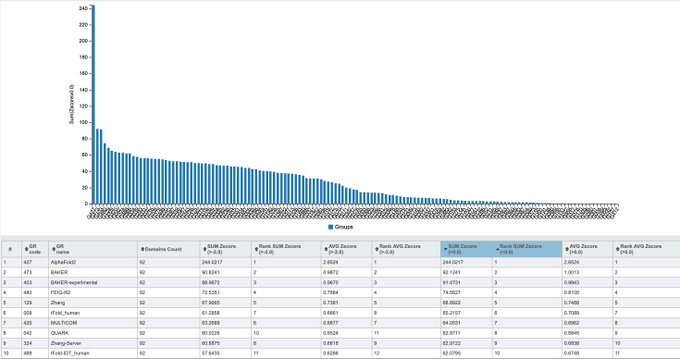
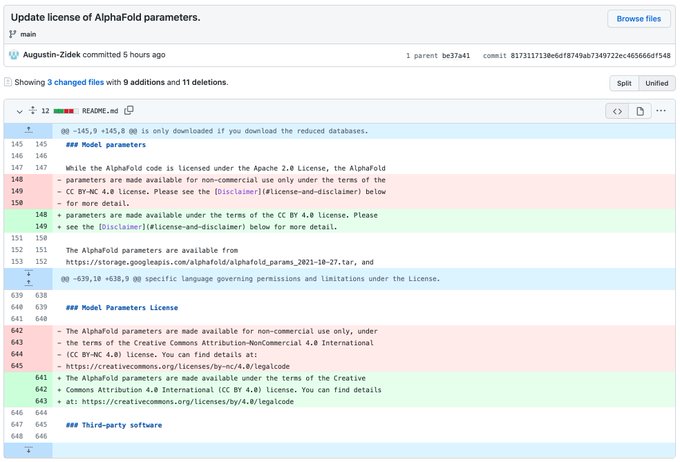
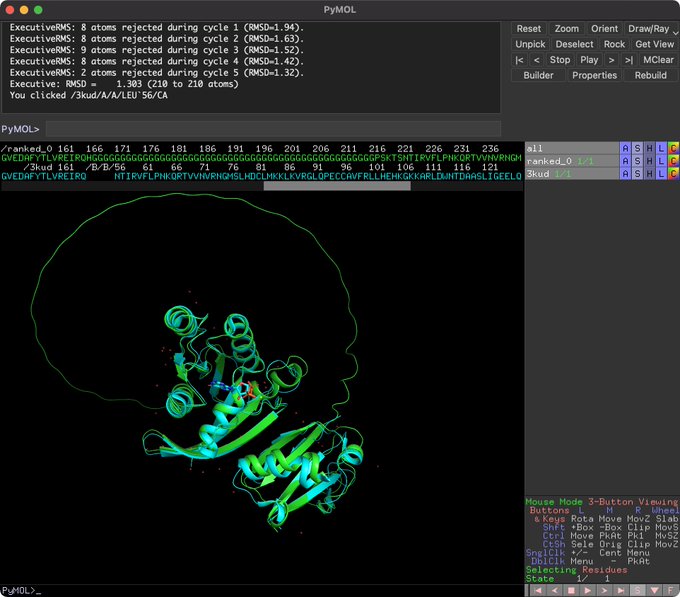
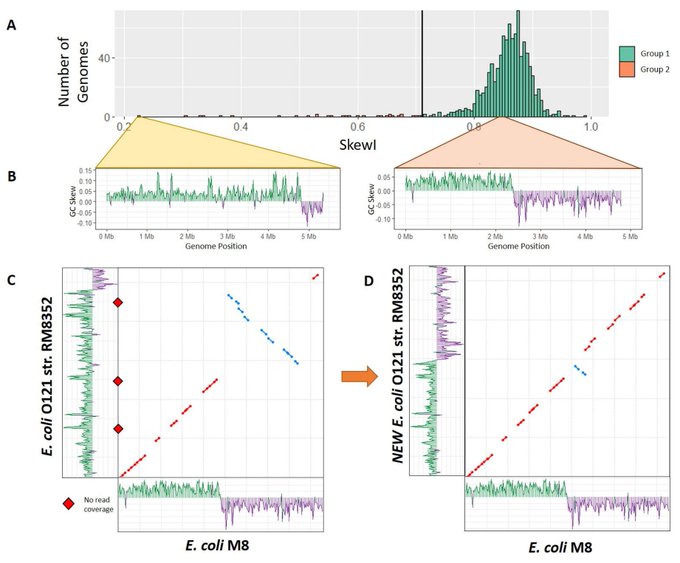
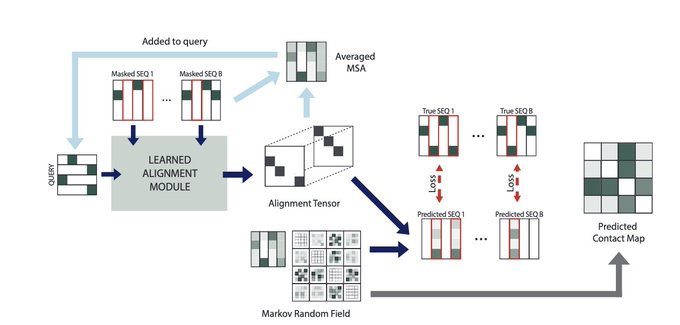
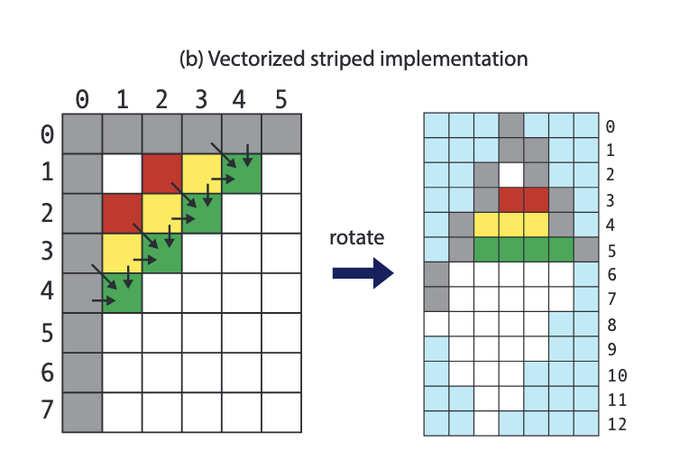
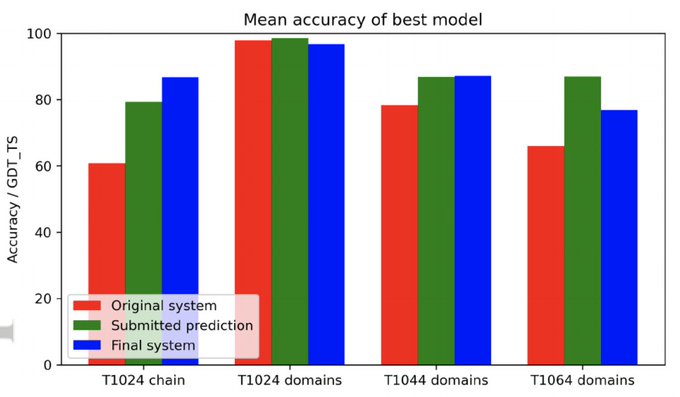
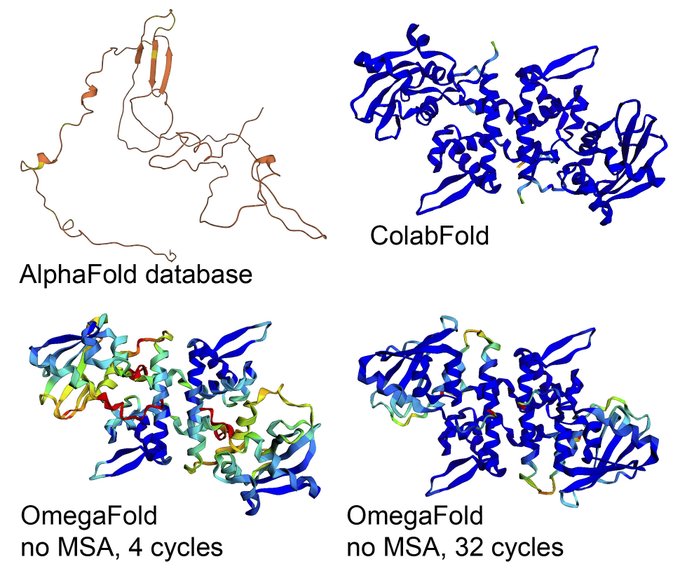
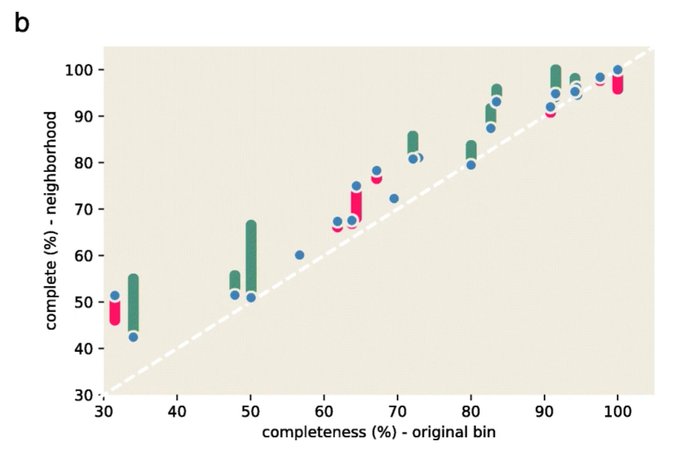
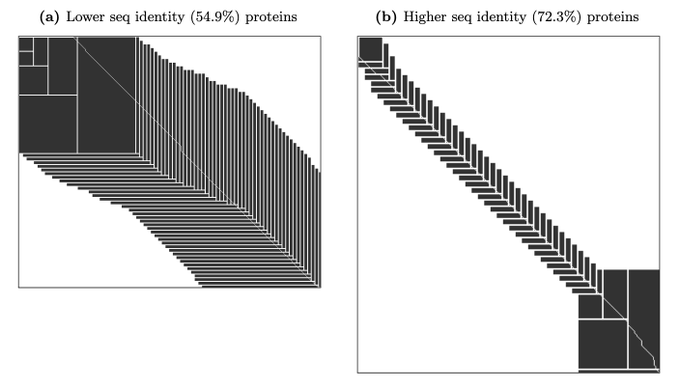
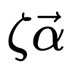Martin Steinegger 🇺🇦
@thesteinegger
5,812
Followers
673
Following
231
Media
1,864
Statuses
Developing data intensive computational methods • PI @ Seoul National University 🇰🇷 • #FirstGen • he/him • Hauptschüler • @martinsteinegger @mstdn .science
Don't wanna be here?
Send us removal request.