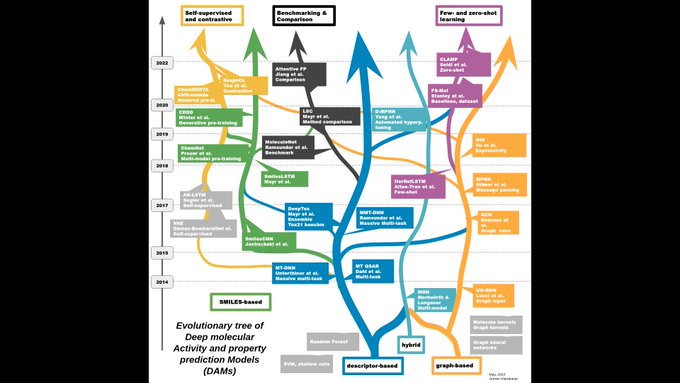
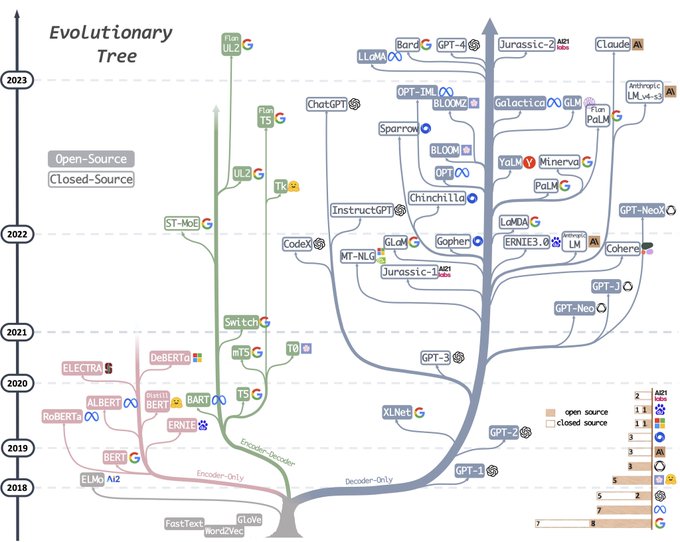
Günter Klambauer
@gklambauer
2,525
Followers
409
Following
95
Media
1,461
Statuses
Deep Learning researcher known for self-normalizing neural networks, applications of Machine Learning in Life Sciences areas; ELLIS program Director
Joined December 2021
Don't wanna be here?
Send us removal request.